

The U.S. Food and Drug Administration’s new Quality Management System Regulation (QMSR) is a historical update that harmonizes 21 CFR Part 820 with ISO 13485:2016 and introduces new terminology, record-keeping structures, and inspection protocols. This marks a pivotal regulatory shift for the medical device industry, aligning U.S. quality system expectations with international standards, making compliance with the international standard a legal requirement rather than a voluntary option.
This change is not just an updated rule; it’s a transformation in how regulatory requirements are defined and enforced. This alignment aims to reduce regulatory burdens for global manufacturers while maintaining safety standards.
The final rule, published in the Federal Register on February 02, 2024, sets an effective date of February 2, 2026. By that time, all manufacturers of finished devices marketed in the U.S. must be in compliance. Read the final rule here.
Why the FDA Is Updating the Quality System Regulation (QSR)
The original QSR, grounded in current good manufacturing practice (cGMP) principles, was first introduced in 1996 under the Federal Food, Drug, and Cosmetic Act (FD&C Act). While foundational, the regulation had not significantly evolved in nearly three decades.
As stated by the FDA:
“The FDA has determined that the requirements in ISO 13485 are, when taken in totality, substantially similar to the requirements of the QS regulation, providing a similar level of assurance in a firm’s quality management system and ability to consistently manufacture devices that are safe and effective and otherwise in compliance with the Federal Food, Drug, and Cosmetic Act (FD&C Act).”
Given this determination, the FDA initiated the rulemaking process that culminated in the QMSR final rule.
The new regulatory framework incorporates ISO 13485 by reference and notes its use by the Medical Device Single Audit Program (MDSAP), a program already utilized by the FDA and four other regulatory bodies from other countries. These updates reflect a larger movement toward simplification and global alignment in medical device quality management.
The FDA cited several motivations for the new requirements in the final rule:
- Reduce duplicative audits and compliance burdens
- Promote consistency with other regulatory authorities and the international standard
- More explicitly incorporate risk management requirements for quality management systems
- Improve patient access to safe, effective devices by accelerating time-to-market
In implementing QMSR, the agency aims to modernize device oversight while maintaining alignment with statutory obligations under the FD&C Act and the quality system requirements that support cGMPs.
What QMSR Means for You: Key Changes
While the QMSR does not upend core quality system expectations, it does reshape how requirements are structured and documented. Here are the most significant updates:
1. ISO 13485 Incorporated by Reference
QMSR formally incorporates by reference ISO 13485:2016, the international standard for medical device quality management systems. This approach allows the FDA to rely on a widely accepted consensus standard while layering in additional QMSR requirements necessary to comply with U.S. law.
Notably, Clause 3 of ISO 9000:2015, Quality Management Systems—Fundamentals and vocabulary, is also incorporated by reference.
2. New Section Structure and Terminology
The new rule significantly reduces written content; from 15 sub-parts (A-O) in the QSR to just two sub-parts (A&B) in the QMSR. Most of the current requirements in Part 820 are withdrawn, replaced by references to ISO 13485:2016.
The QMSR transformation is radical in scope. Many sections have been marked as "Reserved" - meaning they are deleted and held for potential future FDA additions. Only a few sections remain active in some form, with four entirely new sections added.
Two subsections have been retained with some modifications:
- § 820.1 - Scope
- § 820.3 - Definitions: The definitions in ISO 13485 and Clause 3 of ISO 9000 are the new baseline for terminology. FDA provides a list of terms and their definitions that are either not used or not defined in the incorporated ISO standards, as well as a list of terms and their definitions that are explicitly superseded by definitions in QMSR.
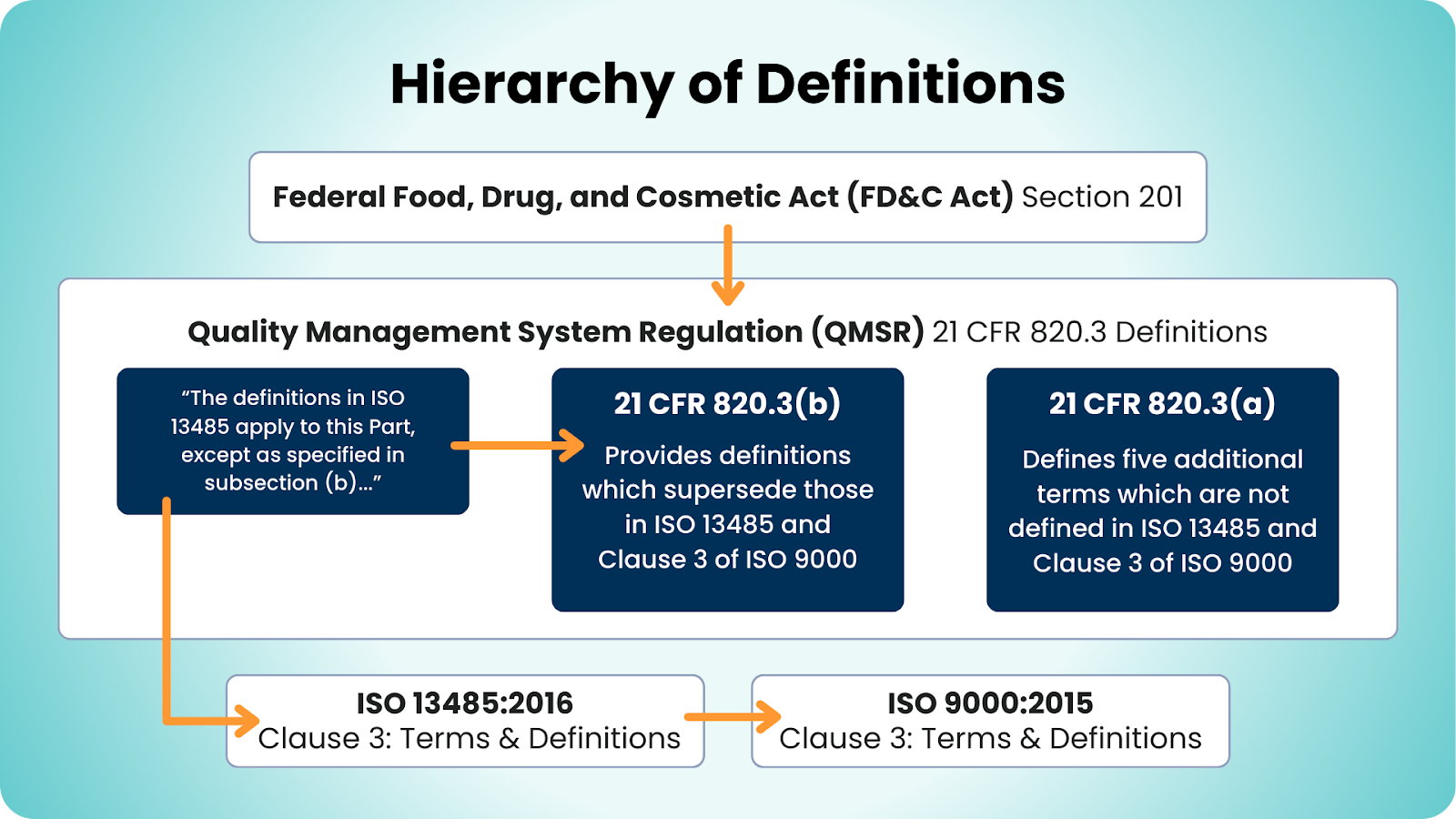
Notably gone are the traditional terms Device Master Record (DMR), Device History Record (DHR), and Design History File (DHF), which are replaced with the unified Medical Device File (MDF) per ISO 13485.
The QMSR introduces four new subsections within 21 CFR Part 820:
- § 820.7 – Incorporation by Reference
- § 820.10 – Requirements for a Quality Management System
- § 820.35 – Control of Records
- § 820.45 – Device Labeling and Packaging Controls
This reorganization also simplifies auditing and internal documentation alignment, making it easier to establish and maintain a single source of truth across all medical device quality documentation.
What the Proposed Rule Told Us and What the Final Rule Made Clear
The FDA’s proposed rule, released in 2022, sparked extensive industry feedback. Many stakeholders raised concerns over terminology changes and documentation impacts. The final rule responded with important clarifications:
- FDA definitions (especially for terms like "device," "labeling," and "combination products") will take precedence where inconsistencies with ISO 13485 exist.
- The removal of DMR, DHR, and DHF does not reduce documentation obligations. Instead, it reinforces the need for consistent, auditable records in the form of the Medical Device File.
- The QMSR includes additional requirements for labeling and packaging activities that extend beyond the scope of ISO 13485 Clause 7.5.
- ISO 13485 certification alone will not exempt manufacturers from FDA inspections. Manufacturers will still be assessed according to the FDA’s updated inspection model, which will revise Quality System Inspection Technique (QSIT) methodologies to align with the new QMSR framework.
For medical device manufacturers, this reinforces the importance of a QMS that is both compliant and flexible, capable of evolving with updates to guidance documents and enforcement practices.
Preparing for the QMSR: How Modern Platforms Make a Difference
The February 2, 2026 effective date is fast approaching, and the transition period offers a vital window for manufacturers to align their systems, language, and documentation with the new rule. However, those using rigid or outdated QMS tools may struggle to adapt to the broader rulemaking scope and shifting expectations.
Here’s where modern, cloud-native QMS platforms become strategic assets. With support for real-time control of records, automated traceability, configurable labeling processes, and robust support for management review and supplier audits, these platforms deliver more than compliance; they enable operational agility.
From centralizing your Medical Device File to supporting cross-functional design controls and enabling continuous improvement through internal audits, the right system can drive both efficiency and inspection readiness.
Moreover, flexible architectures allow teams to respond quickly as the FDA releases additional guidance documents or clarifies expectations in future updates to the federal register.
Final Thoughts: Regulatory Change Is an Opportunity to Strengthen Quality
This is not a minor update; it’s a regulatory reset.
Expect to see increased scrutiny around device labeling, UDI traceability, and packaging controls, especially during FDA inspections. These specific requirements reflect the agency’s intensified focus on downstream risk management across the product lifecycle.
The companies that respond proactively to new and future changes to FDA regulations will be the ones best positioned to meet inspection readiness, reduce risk, and streamline global market access.
The complexity of harmonization requires more than just regulatory awareness; it demands systems built for adaptability. Propel’s cloud-native QMS solution gives medical device manufacturers the control, transparency, and speed needed to stay ahead of shifting regulations.
Whether you're still planning or already implementing updates to your existing quality system, Propel offers a future-ready platform built to help you meet every requirement, old and new.
Propel Software is ready to be your compliance partner. In the coming months, we’ll be publishing further guidance on how to navigate this shift, including practical strategies for updating your QMS requirements and preparing for upcoming FDA inspections.
Need help future-proofing your QMS?
Our team is here to support your transition to the new QMSR without slowing your business down. Reach out today.






